
Cancer Predisposition Program
Our program provides individualized care and counseling to all patients and families affected by cancer predisposition syndromes by harnessing the power of precision medicine and the most up-to-date research from the medical genetics and hematology/oncology community.
View Our Rankings
A pediatric cancer predisposition syndrome happens when a child is born with a genetic variant that changes how a specific gene works. This gene, often called a tumor suppressor gene, cannot protect cells from becoming cancerous when it isn’t working properly. The mutation increases the overall risk for affected children to develop cancer or a number of benign tumors, especially at younger ages.
The Cancer Predisposition Program at the Aflac Cancer and Blood Disorders Center cares for patients who have this increased risk, from birth, through childhood and into adolescence.
Cancer predisposition syndromes cause about 10 percent of all pediatric cancers. Diagnosing one of these syndromes leads to proactive and targeted surveillance of the patient and identification of other at-risk relatives.
Read Stories of Hope and Inspiration From Our Patients
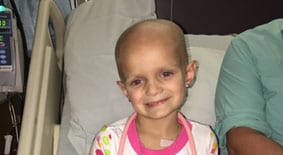


The Cancer Predisposition Program treats children under the age of 21 who are at an increased risk of developing cancer due to a cancer predisposition syndrome. Below are some of the most common cancer predisposition syndromes found in children.
CMMRD is a genetic condition in which individuals are born with a predisposition to develop many different types of childhood and adult-onset cancers. The average age of cancer diagnosis in patients with this syndrome is age 7.
Cancers most often diagnosed in children with CMMRD include:
- Leukemia and lymphoma (especially non-Hodgkin lymphoma)
- Glioblastoma multiforme and high-grade gliomas
- Colon cancer
CMMRD syndrome is caused by genetic changes in both copies of a mismatch repair gene. These include MLH1, MSH2, MSH6, PMS2 and EPCAM. An individual must inherit a nonworking copy of a gene from each of his or her parents in order to be affected by this syndrome. In most cases, the parents themselves will not be diagnosed with cancer; the child with cancer is most often the first patient identified in the family. Each parent of a child with CMMRD has an adult-onset syndrome called Lynch syndrome, which increases lifetime risk of colon, endometrial and other types of cancers.
The incidence of CMMRD syndrome is unknown and thought to be very rare. Fewer than 500 patients with CMMRD syndrome have been reported in the medical literature.
CMMRD syndrome can be diagnosed through genetic testing that is facilitated by a genetic counselor or genetics services provider.
Children and adults with CMMRD syndrome should receive care at centers with expertise in this syndrome.
DICER1 syndrome, also known as DICER1-pleuropulmonary blastoma familial tumor predisposition syndrome, is a condition in which an individual is born with genetic changes that predispose him or her to develop benign and malignant tumors during childhood, through adolescence and, rarely, as an adult.
In childhood, these most often include:
- Pleuropulmonary blastoma
- Ovarian sex cord stromal tumors
- Cystic nephroma
- Genitourinary rhabdomyosarcomas
- Increased risk for familial nodular goiter
- Pineoblastomas
- Pituitary blastomas
- Ciliary body medulloepitheliomas
- Renal sarcomas
The genetic changes that cause this syndrome are found in the DICER1 gene, a tumor suppressor gene. When working normally, both copies of this gene help suppress the development of cancer. However, individuals with DICER1 syndrome are born with one working copy and one nonworking copy.
This genetic change is passed down from either parent 80% of the time. However, 20% of the time, this genetic change occurs on its own in an oocyte or sperm cell prior to fertilization.
DICER1 syndrome can only be diagnosed after genetic testing. This process is facilitated by a genetic counselor, who will discuss family history, complete a risk assessment, and explain testing benefits and options.
Not all individuals with a diagnosis of DICER1 syndrome will develop tumors or cancer. Surveillance strategies are important because if cancer does develop, it can be caught and treated as early as possible.
FAP syndrome is a genetic syndrome that predisposes individuals to develop hundreds to thousands of adenomatous polyps, in addition to colorectal cancer, papillary thyroid cancer and other cancers. Every patient with FAP will develop colorectal cancer, typically by age 40, unless screening and preventive measures are taken.
In childhood, patients with FAP are at risk for developing:
- Hepatoblastoma (a type of liver cancer), typically before age 5
- Medulloblastoma (a type of brain cancer)
- Thyroid cancer
- Gastrointestinal (especially colorectal) polyps
- Desmoid tumors (typically noncancerous)
- Extra/additional teeth
FAP is caused by inherited genetic changes in the APC gene. A child of a parent diagnosed with FAP has a 50% chance of inheriting this condition. While the majority of cases are passed down to a child from a parent, nearly 20% of cases of FAP occur for the first time in the affected child and are not inherited.
The incidence of FAP is estimated to be 1 in 14,000.
FAP can be diagnosed through genetic testing, which is typically performed by a genetic counselor. Family history of polyposis, “carpeted colon” appearance on a colonoscopy or colon cancer diagnosed at an early age (20 to 40 years old) can also increase the likelihood of this diagnosis.
Guidelines and recommendations for the care and surveillance of children and adults with FAP include:
- Annual colonoscopy screening beginning between ages 10 and 15.
- Cervical palpation of thyroid.
- Abdominal ultrasound and labs.
- Annual physical exam by pediatric hematologist/oncologist with expertise in caring for patients with FAP.
HPPS includes a number of different genetic syndromes that predispose individuals to develop paragangliomas (tumors that form from neuroendocrine tissue) and pheochromocytomas (similar tumors that form in the adrenal medulla). These tumors may or may not secrete hormones, and while the majority are benign (nonmalignant), a few of them can go on to become cancerous (malignant).
Patients with HPPS are also at an increased risk of developing the following, depending on the specific genetic syndrome:
- Renal cell carcinoma (a type of kidney cancer)
- Gastrointestinal stromal tumors
- Papillary thyroid carcinoma, pituitary adenomas and other neuroendocrine tumors
HPPS is caused by genetic changes in the following genes, which act as tumor suppressor genes:
- SDHx (SDHA, SDHB, SDHC, SDHD, SDHAF2)
- TMEM27
- MAX
- VHL
- NF1
- RET
A tumor suppressor gene, when working properly, encodes proteins that prevent the growth and development of tumors in the human body. The incidence of HPPS is unknown.
HPPS can be diagnosed through genetic testing that is facilitated by a genetic counselor or genetics services provider. Any patient diagnosed with a paraganglioma or a pheochromocytoma should be referred to genetic counseling or medical genetics to discuss genetic testing and be evaluated for HPPS.
The medical monitoring of children and adults with HPPS should start between ages 6 and 8 and often includes:
- Annual lab work (e.g., plasma metanephrines).
- Whole-body MRIs (skull to pelvis) once every two years.
- Blood pressure checks at all clinical visits.
- Physical exam (annually).
LFS is a syndrome that predisposes individuals to develop several types of benign and malignant tumors. In childhood, this most often includes:
- Choroid plexus carcinoma, a type of brain tumor
- Adrenocortical carcinoma, which is present in the adrenal glands located on top of the kidneys
- Osteosarcoma, or bone tumor
- Rhabdomyosarcoma, or soft tissue tumor
LFS is caused by genetic changes that occur in the TP53 gene, which is often called the guardian of the genome and has several vital functions, including preventing tumor development and controlling the cell cycle. In 7-20% of cases, LFS may occur on its own, without being passed down from either parent. However, in most cases, genetic alterations, or changes in the TP53 gene that cause LFS, are inherited from one parent. This means that a patient’s full siblings also have a 50% chance of having Li-Fraumeni syndrome.
Not everybody with LFS will develop cancer in childhood, though almost 100% of women and 85-90% of men with this syndrome will go on to develop cancer in their lifetimes. In large groups of patients with LFS, 22% received a cancer diagnosis by age 5, and 40% developed their first cancer by age 18.
Diagnosis of LFS occurs through genetic testing, which is often facilitated by a genetic counselor or a clinical oncology team.
Due to the increased risk of cancer, monitoring is recommended for both children and adults with LFS. This is best done at a center where physicians, genetic counselors and geneticists have expertise in LFS.
A diagnosis of LFS may change treatment plans for existing cancers, as well as medical management and future surveillance or monitoring plans, as individuals with LFS are extremely sensitive to radiation and at increased risk for secondary cancers.
Research has shown that patients with LFS who undergo regular surveillance have a significantly higher overall survival than those who do not undergo regular surveillance.
Once an individual has been identified as having LFS, a genetic counselor can help identify family members who may be at risk for LFS. All family members at risk may choose to pursue genetic counseling and testing.
Rhabdoid predisposition syndrome is a genetic condition in which individuals are born with an increased likelihood of developing soft tissue tumors called rhabdoid tumors. These develop primarily in the kidneys and brain but can also develop elsewhere in the body, such as in the ovaries. In the brain, they are called atypical teratoid rhabdoid tumors.
Most often, these tumors are diagnosed before a child’s fifth birthday, but they can also present in older individuals.
This syndrome is caused by genetic changes in the SMARCA4 and SMARCB1 genes. Both of these genes act as organizers of the genome, specifically with regard to chromatin structuring and packing, or chromatin remodeling.
Individuals with rhabdoid predisposition syndrome are born with these changes, and in about 40% of cases, the changes are passed down from one parent. Not everybody born with rhabdoid tumor predisposition syndrome will go on to develop these types of tumors.
Rhabdoid predisposition syndrome can be diagnosed through genetic testing, which is often facilitated by a genetic counselor or genetics services provider.
Guidelines for monitoring of children and adults with rhabdoid predisposition syndrome include:
- Brain MRI every three months until age 5.
- Ultrasound of abdomen every six months.
- Physical exams by pediatric hematologist/oncologist once a year.
VHL syndrome is a genetic condition in which individuals are born with a predisposition to develop a range of benign and malignant tumors. These include:
- Renal cell carcinoma (a type of kidney cancer)
- Tumors of the liver, pancreas and adrenal gland (pheochromocytomas)
- Hemangioblastomas (brain and spine) and retinal angiomas
- Endolymphatic sac tumors that may cause hearing loss
- Epididymal and broad ligament cystadenomas
VHL syndrome is caused by genetic changes in the VHL gene, which acts as a tumor suppressor. A tumor suppressor gene, when working properly, encodes proteins that prevent the growth and development of tumors in the human body.
The incidence of VHL syndrome is estimated to be 1 in 30,000.
VHL syndrome can be diagnosed through genetic testing that is facilitated by a genetic counselor or genetics services provider.
Some families have had a diagnosis of VHL syndrome without any previous genetic testing, based on multiple hemangioblastomas and other tumor types that are associated with this syndrome.
Guidelines for the medical monitoring of children and adults with VHL syndrome often include:
- Annual lab work (e.g., plasma metanephrines).
- Annual eye exams by an ophthalmologist familiar with this syndrome.
- Biannual brain, spine and abdominal MRIs.
- Blood pressure checks at all medical visits.
At the Cancer Predisposition Program, our goal is to provide individualized care and counseling to all patients and families affected by cancer predisposition syndromes by harnessing the power of precision medicine and the most up-to-date research from the medical genetics and hematology/oncology community.
During a typical new patient appointment at our clinic, we provide:
- Risk assessments for cancer predisposition syndromes through collection of personal and family health history.
- In depth genetic counseling before and after genetic testing, including education on implications of diagnosis for both patient and family members.
- Comprehensive medical evaluations by a board-certified pediatric hematologist-oncologist.
- Personalized tumor surveillance options based on and the current guidelines and each family’s needs.
- Opportunities to participate in research.
What should you bring to your child’s appointment at the Cancer Predisposition Program?
- Any previous genetic testing results.
- Knowledge about the family history of cancer (in child, siblings, parents, aunts and uncles, and grandparents), including type(s) and age of diagnosis.
- Any questions or concerns regarding the risk of cancer in your child and/or family members.
Building a pedigree
The cancer predisposition team will build a pedigree, or medical family tree, for your child or teen based on the family history and genetic testing information provided at the first appointment. A pedigree is a graphic representation of a family’s medical history and genetic relationships as it relates to cancer, typically going back three generations. A pedigree takes into account:
- Who in the family has been diagnosed with cancer.
- How old they were when the cancer was diagnosed.
- What type of cancer they had.
Building a pedigree allows the cancer predisposition team to look for certain patterns, which helps the team in its risk assessment. Certain patterns increase the likelihood of a cancer predisposition syndrome. These patterns include:
- Cancer types
- Age of diagnosis
- Previous results from cancer screening (e.g., history of colon polyps)
- Any previous lumps/bumps (e.g., thyroid nodules)
- Side of the pedigree affected (maternal, paternal or both)
What is genetic counseling?
Genetic counseling helps patients understand how genetic conditions might affect them and their family members.
All new patients referred to the Cancer Predisposition Program will meet with one of our team’s genetic counselors. The genetic counselor will take a personal and family history, perform a risk assessment, describe any relevant cancer predisposition syndromes, recommend surveillance and coordinate genetic testing, if applicable.
If the patient or a family member has had previous genetic testing, it is helpful to bring the report to the appointment.
Our Cancer Predisposition Program takes a multidisciplinary approach by incorporating comprehensive, pediatric-focused genetic counseling and cutting-edge genetic tools to ensure the highest level of care for each patient and family.
Program highlights
- U.S. News & World Report ranks the Aflac Cancer and Blood Disorders Center among the top pediatric cancer programs in the country.
- We are the only pediatric cancer predisposition program in Georgia.
- We provide services to more than 300 patients and family members who are at risk for developing cancer.
- More than 340 clinical studies available for patients, 28 Aflac investigator-initiated clinical trials, and over 675 study enrollments in 2022.
Dr. Porter receives St. Baldrick’s Consortium Research Grant
Christopher Porter, MD, received a $2.5M grant to create a Consortium for Childhood Cancer Predisposition aimed at improving outcomes for those most at risk of cancer through better identification, early tumor detection, optimized psychosocial support and cancer prevention.
READ MOREPatients must be referred to our Cancer Predisposition Program. The following are among the reasons for referral to the program:
- The patient or a blood relative has a known cancer predisposition syndrome (e.g., Li-Fraumeni syndrome).
- Tumor type is suggestive of a specific cancer predisposition syndrome (e.g., adrenocortical carcinoma, renal cell carcinoma or pleuropulmonary blastoma).
- Tumor genetic testing is suggestive of a cancer predisposition syndrome (e.g., TP53 mutation identified in osteosarcoma tissue biopsy).
- Two or more malignancies are found in one patient (e.g., patient diagnosed with rhabdomyosarcoma and a subsequent osteosarcoma).
For referring physicians
If you are a physician and you have questions regarding a referral for a patient to the Cancer Predisposition Program, please complete the steps outlined below:
Fax the following to our scheduling team:
- Patient demographics sheet, including insurance information.
- Reason for referral (briefly summarize why patient needs to be seen at the Cancer Predisposition Program).
- Any genetic testing records (either the patient’s or the parents’), as well as any history of cancer diagnosis (in patient, in patient’s siblings or in patient’s parents).
Referral contact:
Cancer Predisposition Program scheduler
Fax: 404-785-3760
Phone: 404-785-3519
Physicians should refer pediatric patients with a known history or family history of the following syndromes:
- Li-Fraumeni syndrome (LFS)
- Familial adenomatous polyposis (FAP)
- Von Hippel Lindau (VHL) syndrome
- DICER1/PPB syndrome
- Multiple endocrine neoplasia Type 1 and 2
- Cowden syndrome
- Peutz-Jeghers syndrome (PJS)
- Juvenile polyposis syndrome
- Rhabdoid tumor predisposition syndrome
- Constitutional mismatch repair deficiency (CMMRD) syndrome
- Hereditary paraganglioma pheochromocytoma syndrome (HPPS)
- Gorlin/NBCCS syndrome
If you are unsure if a referral is appropriate, email genetic counselor Bojana Pencheva, MMSc, CGC.
1. Family history of the child with cancer:
- Two or more malignancies at childhood age (younger than 18 years of age)
- A first-degree relative (parent or sibling) with cancer under age 45
- Two or more second-degree relatives with cancer under age 45 on same side of family
- The parents of the child with cancer are related (consanguinity)
2. A person with one or more of these tumors in childhood:
- Adrenocortical carcinoma
- Atypical teratoid rhabdoid tumor (ATRT)
- Basal cell carcinoma
- Cerebellar gangliocytoma (COLD syndrome)
- Choroid plexus carcinoma
- Cystic nephroma
- Desmoid tumors
- Endolymphatic sac tumors (ELST)
- Gastrointestinal stromal tumors (GIST)
- Gonadoblastoma
- Hemangioblastoma
- Hepatoblastoma
- Juvenile myelomonocytic leukemia
- Low hypodiploid acute lymphoblastic leukemia
- Malignant peripheral nerve sheath tumors
- Medullary thyroid carcinoma
- Medulloblastoma
- Melanoma
- Meningioma
- Neuroblastoma
- Optic glioma
- Osteosarcoma
- Sertoli-Leydig cell tumor
- Small cell carcinoma of ovary
- Pancreatic islet cell tumor
- Paragangliomas/pheochromocytomas
- Parathyroid carcinoma
- Pleuropulmonary blastoma
- Pituitary blastoma
- Pineoblastoma
- Plexiform neurofibroma (NF1)
- Retinoblastoma
- Rhabdomyosarcoma (anaplastic)
- Rhabdoid tumors (especially renal and liver)
- Renal cell carcinoma
- Schwannoma
- Spinal cord ependymoma
- Subependymal giant cell tumor
- Wilms’ tumor (bilateral only)
- Or a cancer of adulthood (e.g., colorectal, ovarian)
3. A patient with two malignancies, one with onset under the age of 18 (unless the second malignancy is consistent in time and/or tissue type with those expected from his or her treatment regimen)
4. A child with cancer and congenital anomalies or other specific symptoms:
- Aberrant growth (large head circumference, hemihyperplasia)
- Skin anomalies (cafe au lait spots, hypersensitivity to sunlight)
- Congenital anomalies, facial dysmorphisms and intellectual disability may warrant a referral to a clinical geneticist for initial workup.
- Christopher Porter, MD
- Sarah Mitchell, MD
- Bojana Pencheva, MMSc, CGC
- Kelsi Hagerty, MMSc, CGC
- Erin Seibel, MMSc, CGC
Contact Us 404-785-1200